|
||||||||||
Computational Chemistry Overview
A Brief Review of Conformational Analysis
An Overview of Computational Chemistry
Unfortunately, Chem3D is no longer available. There is a 3D app for iPad that you can try using. See the Courseware section for the download link.
There are two versions of Chem3D in general use: Ultra 11 version which you might have downloaded and are using on your personal computer and Pro 10 version that is installed on the computers in the GMU classroom and my lab computer. They perform very similarly even though the interfaces appear different.
All quizzes in using Chem3D necessarily refer to version 10. Assignments may incorporate aspects of Ultra 11.
Molecular Modeling with CHEM3D ver. 10
Conformational Analysis of Cyclohexane and Cyclohexanols
Conformational Analysis of n-Butane: Combined Chem3D and Excel
Overlaying Two Molecules in the Last Class Assignment
After copying and pasting two molecules in one window, open the Model Explorer window. You will see "Fragments" listed that correspond to the two molecules (confirm by selecting one and then the other Fragment). If you have lone pairs showing on the model, they will also be listed as Fragments. You can delete these by selecting the Fragments in the Model Explorer and deleting them.
There might be occasion in the classroom to use the Overlay calculation in Pro 10, which is a little less efficient to use than the Ultra 11 Fast Overlay that you use in the Protein Modeling assignment. Here are instructions for performing an Overlay in Pro 10:
Display in the 3D window the two molecules that are to be overlaid. Select pairs of atoms that will be used in the overlay attempt. First choose an atom in molecule 1, and hold down the Shift key while selecting the complementary atom in molecule 2. Choose Structure | Measurements | Display distance. Then select a second pair of atoms and display the distance. At least three pairs must be selected. Then choose Structure | Overlay | Minimize. [This would not be part of a quiz.]
You can download DS Visualizer by clicking on the links in the CourseWare section of the Chemistry 350 web site.
The DS Visualizer version now available is much less friendly to use than the instructions below indicate.
DS Visualizer allows you to examine the 3D structure of molecular models. All the common structure file formats can be opened and saved. The most important for molecular modeling purposes are those for large molecules such as proteins and nucleic acids (pdb) and for general molecule viewing of smaller molecules (mol). DS Visualizer will also allow you to save molecular "poses" so that the file (msv) opens with the display options you chose instead of a default display. The graphics file formats jpeg, bmp, and png are supported.
By default, the Visualizer opens with a Files window on the left and an empty 3D window on the right. You can close the Files window. Also by default, drag-and-drop capability is enabled and you can load multiple files in the 3D window.
Many of the menu items are familiar as typical Windows menus or are largely self-explanatory. You should explore them with a molecular model open in the window. The tool bars are detachable and you can move them. The program comes with extensive sample files.
Display Styles
Opening a file in the 3D window will result in a line (wireframe) display of the molecule. The other styles available are stick, ball and stick, and CPK (spacefilling -- shown below). Proteins have additional styles. The default colors are by element (oxygen = red, nitrogen = blue, etc.). To change the display, choose View | Display style... or right-click the mouse and choose Display style...
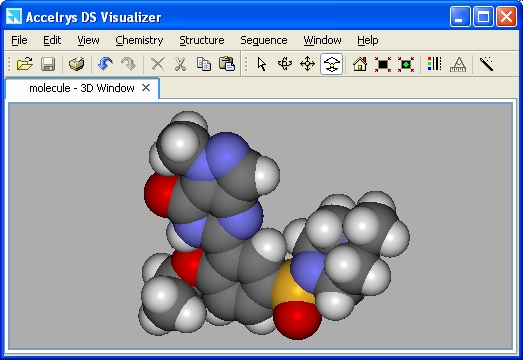
It is often advantageous not to show hydrogen atoms in the display. Choose Chemistry | Hydrogens | Hide.
Other viewing options that you can change may be chosen under Edit | Preferences... | 3D Window.
Choose Ultra-high Quality and make sure Fast render on move is not selected (to see why, rotate the molecule with and without the box checked).
You can also change the Background Color.
Be sure the box "Enable graphics hardware acceleration" is not checked.
Manipulating
Structures
 |
One or more atoms, bonds, or molecules can be selected using the Selection tool. To select one item, left-click on it. To select more than one item, press [Shift] and left-click simultaneously. An entire molecule, or parts of it can be selected by holding down the left mouse button and dragging the selection tool around the display or pressing [Ctrl]A. To rotate a molecule, click the rotation tool in the toolbar. To rotate around the X or Y axis, depress the left mouse button, hold and drag. To rotate around the Z axis, hold the [Shift] key down, depress the left mouse button and drag horizontally in the window. To rotate one of two or more structures in a window, select the structure, hold down the [Ctrl] key and rotate as above. |
The Translation and Zoom tools operate similarly to the rotation tool except there is no Z-translation or zoom.
Multiple Molecules
You can have more than one molecule open in a window by dragging and dropping the file into the open 3D window.
You can also open each molecule in separate windows. In one window, choose Edit | Copy. In the other window, choose Edit | Paste. Sometimes the pasted molecule is not visible although it is still selected. Without clicking in the window, choose the translation tool, hold down the [Ctrl] key and drag around the window until the selected molecule is visible. When it is properly placed in the window, click outside the selection.
You can also open a Hierarchy window to select one or more molecules (see below). You can set the display styles individually and differently for each molecule or part of a molecule. Set a display style for all atoms. Then select atoms or bonds that you want to change and set the display style for them only.
Windows
3D Window
The 3D window is the default model display window in which a molecular
model is open, moved, rotated, etc.
Hierarchy
Window -- Displaying Atoms and Objects
A Hierarchy window shows the organization of atoms and objects
in a molecule. All atoms and bonds in the molecule are listed.
(To show the bonds, right-click in the Hierarchy window and check "Display bonds".)
If the molecule is a protein, the amino acid residues are listed
with their constituent atoms in a sub-listing. Selections of
single or multiple atoms can be made in either the Hierarchy
or 3D window.

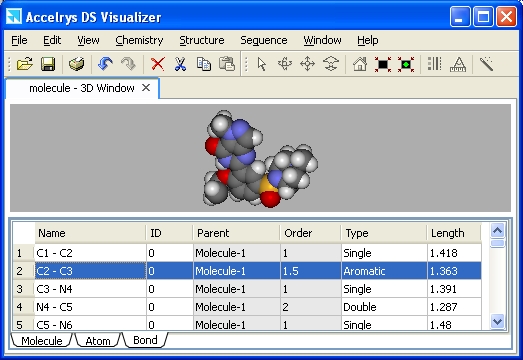
Data Table
Window
The data table contains
additional property information about the atoms, bonds, and other objects
associated with the molecule(s). Objects can be selected in both the data
table and the 3D window by clicking on the relevant row numbers in the
table.
To copy table data to paste in Excel, select and copy the relevant table cells. To sort column data in the opposite order, double-click on the column heading.
Saving Image Files
To save the molecule in the 3D window as an image, choose File | Save As... | Image Files. Unlike many programs, you need to type the extension in order to save in the desired format. The default format is .png -- do not choose this format. Instead, choose a file name and the bmp format for the best quality graphic. If you want to change to another format later, such as jpg, do it in a graphics program.
A dialog window will open asking you for the width and height of the image. Sizing the 3D window automatically changes the size. Or if you have a full-size window open, you can change the size of the saved image in the dialog window.
Drawing Molecules in DSV
To access the drawing tools, choose View | Toolbars | Sketching. When you are finished, choose Structure | Clean Geometry. This will adjust the bond lengths and angles to more optimum values.
It is necessary sometimes to adjust the charge, hybridization, or bonding in a molecule. Here are some instructions for fixing molecules. Here are some practice problems.
This is a brief document about amino acids and proteins.
Torsion Tool
After drawing a molecule in DSV, you might want to make some adjustments to the dihedral angles. From the Sketching tool bar, choose the Torsion tool. Click on a bond and while holding down the mouse button, drag in the window to change the angle. There is a practice problem in the next section.
Hydrogen Bond Monitor
When there is an internal hydrogen-bond in a molecule (or many), you can display it as a dotted line. Choose Structure | Monitor | Hbonds. The default cut-off distance for the hydrogen-bond is 2.5 A. You can change the cut-off by selecting "Hbond Monitor" in the Hierarchy window, right-clicking and then choosing "Attributes of Hbond Monitor". Increase the distance to find other potential H-bonds.
You can examine the effect of changing a dihedral angle on the formation of an intramolecular hydrogen-bond. Here is a molecule to practice with: H2NCH2CH2CH2OH. When you first invoke the H-bond monitor, you will see one where the OH is the HBD and N is the HBA. Rotate the C-OH bond so that the H-bond disappears. Then rotate the C-NH2 bond until the NH2 is the HBD and O is the HBA. Change the cut-off distance to see the effect of rotation.
Intermolecular hydrogen bonding can be seen in this file of two methanol molecules.
Structure of a Mol file
Behind the 3D display of a molecule in any molecular viewer is a simple text document. Here is a simplified explanation of the text for a .mol file. Open a text editor such as TextPad (better than Notepad) and then open any of your mol files.
{kind=link}