Molecular Modeling
Alex Strube
In this assignment, I downloaded a model of a protein-heterocompound
complex involving aspirin as the
heterocompound, in .pdb format, from RCSB PDB, a protein database. I
extracted the model of the complexed aspirin from the larger model of the
complex using DS Visualizer software. It is displayed as Figure 1.
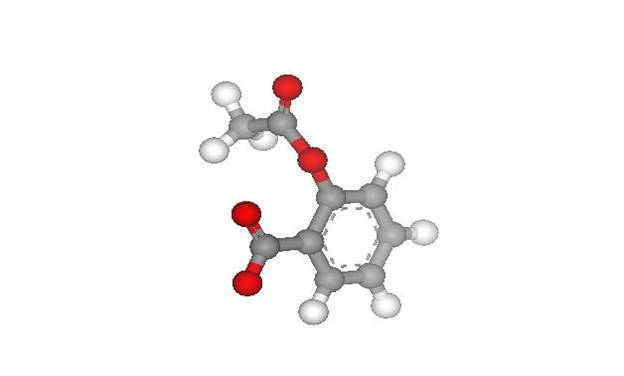
Fig. 1: Ball and Stick model
of aspirin (AIN)
Figure 2 is a Lewis structure of aspirin drawn by me using ChemSketch
software. It includes free pairs of electrons.
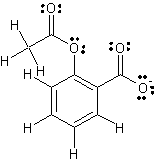
Fig. 2: Lewis structure of aspirin
(AIN)
Figure 3 is the complex, with the
protein part displayed as solid ribbons. The oxygens in the heterocompound are
red.
Fig.
3: Complex formed between group II phospholipase A2 (1tgm) and aspirin
(AIN).
PART
2
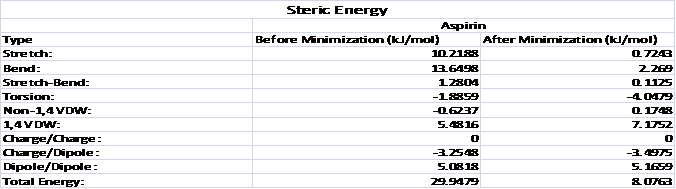
Table 1: single-point energy calculation on
the extracted heterocompound and energy minimization
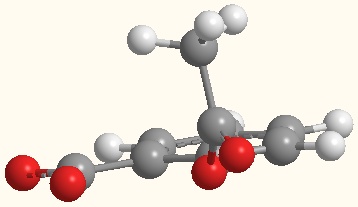
Fig.4: Aspirin in the conformation in which it complexes
with 1tgm.

Fig. 5: Energy minimized aspirin conformation.
Table shows steric energy values for aspirin
in the conformation in which it complexes with 1tgm compared to its minimum
energy conformation. The value for total steric energy for the original
conformation was 29.9479 kJ/mol; The value for total steric energy for the
minimized conformation was 8.0763 kJ/mol. For stretch energy, which
represents the energy
associated with any bonds that are distorted from their optimal length, the value for the original conformation
was higher than the value for the minimized conformation.
For bend energy, which represents
the energy associated with any bond angles that are deformed from their optimal
values, the value for
the original conformation was higher than the value
for the minimized conformation. For stretch-bend energy, which represents the energy required to
stretch the two bonds involved in a bond angle when that bond angle is severely
compressed, the value
for the original conformation was higher than the value for the minimized conformation. For torsion energy, which represents the energy associated with
deforming torsional angles (dihedral angles) in the molecule from their ideal
values, the value for
the original conformation was higher than the value
for the minimized conformation was. For Non-1,
4 van der Waals energy, which represents the energy for the through-space interaction
between pairs of atoms that are separated by more than three atoms, the value for the original conformation
was lower than the value for the minimized conformation was.
For 1, 4 van der Waals energy, which
represents the
energy for the through-space (non-bonded) interaction of atoms separated by two
atoms, the value for the
original conformation was lower than the value
for the minimized conformation was. For dipole/dipole energy, which represents the energy
associated with the interaction of bond dipoles, the value for the original conformation
was lower than the value for the minimized conformation was.
Fig. 6: The original heterocompound conformation
superimposed with the energy minimized conformation.
The original heterocompound conformation was superimposed
with the energy minimized conformation using the overlay function of Chem3D
Ultra. Use this function by going to View
| Model Explorer:
A pane identifying the each molecule in the active window as fragments will
open. Choose one fragment using Structure | Overlay | Set
Target Fragment. Select the other
fragment and choose Structure | Overlay | Fast Overlay. The molecules will be superimposed.
Except for a
few hydrogen atoms, the atoms of the two conformations all partially overlap.
Perhaps this means the original conformation was already at a low energy.
PART 3

Fig. 7: Primary structure of 1tgm.

Fig. 8: Ligplot for AIN.

Fig.8: Ligand Interactions with Certain Amino Acids on 1tgm.

Fig.9: Ribbon Model of 1tgm Showing Legend Interactions with
Certain Amino Acids on 1tgm.
Figure 7 shows the primary structure
of Phospholipase A2, also known by the PDB ID: 1tgm. Each letter represents an
amino acid; the red dots over some letters indicate ligand interactions.
Figures 8, 9, and 10 show the interaction
between aspirin and three amino acid residues, Leu 2, Ala 18, and Ile 19, in
the Phospholipase A2 protein. Figure 8 is a LigPlot. The “eyelashes” in figure
8 indicate hydrophobic interactions between these amino acid residues in the
protein and the heterocompound aspirin.
PART 4
|
Amino acid residue
& position #
|
Atoms in hetero
that interact
|
!interaction
|
|
LEU 2
|
C8 & C9
|
hydrophobic
|
|
ALA 18
|
C4, C3, C7, O1 & O2
|
hydrophobic
|
|
ILE 19
|
C4, C3, C7, O1 & O2
|
hydrophobic
|
Table 2: Amino Acid Residue
Interactions.
Table 2 lists the amino acid residues in 1tgm
that interact with the complexed heterocompound aspirin, their positions in the
protein’s primary structure, which atoms they interact with in the
heterocompound, and what type of interaction (hydrogen bond or hydrophobic) the
LigPlot indicates.
References:
1. RCSB. “1tgm.” RCSB
PDB: Structure Explorer. Dec. 2,
2008. http://www.rcsb.org/pdb/explore/explore.do?structureId=1TGM
2.Singh,
N., Jabeen, T., Sharma, S., Bhushan,
A., Singh, T.P.  Crystal
structure of a complex formed between group II phospholipase A2 and aspirin at
1.86 A resolution To be Published. Similar published article link: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=2pws&template=main.html.
Crystal
structure of a complex formed between group II phospholipase A2 and aspirin at
1.86 A resolution To be Published. Similar published article link: http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=2pws&template=main.html.
3. PDBSum. “Ligand/Metal
Interactions: 1tgm.” http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=1tgm&template=ligands.html&l=1.1